If you’re a regular at From Atoms To Words, you won’t be surprised to hear that I’ve always been intrigued by atoms and molecules, a fascination that led me down the path of quantum chemistry. During my scientific career, I’ve delved into the study of systems of biological relevance, such as small molecules interacting with DNA. One area I would have loved to explore more is enzymes, the catalytic powerhouses of living organisms. The closest I got was one of my last major projects, which focused on photosystem II, a protein crucial in photosynthesis, nature’s method of turning sunlight into energy. Using quantum chemistry, we studied its structure and what we found was pretty surprising. It’s a piece of work I’m really proud of. Now, wait a minute! Can we really use quantum chemistry to study enzymes and enzymatic reactions? Well, that’s exactly what we’re exploring today, with the cluster approach – a key method in quantum chemistry. Ready to get started? Let’s go.
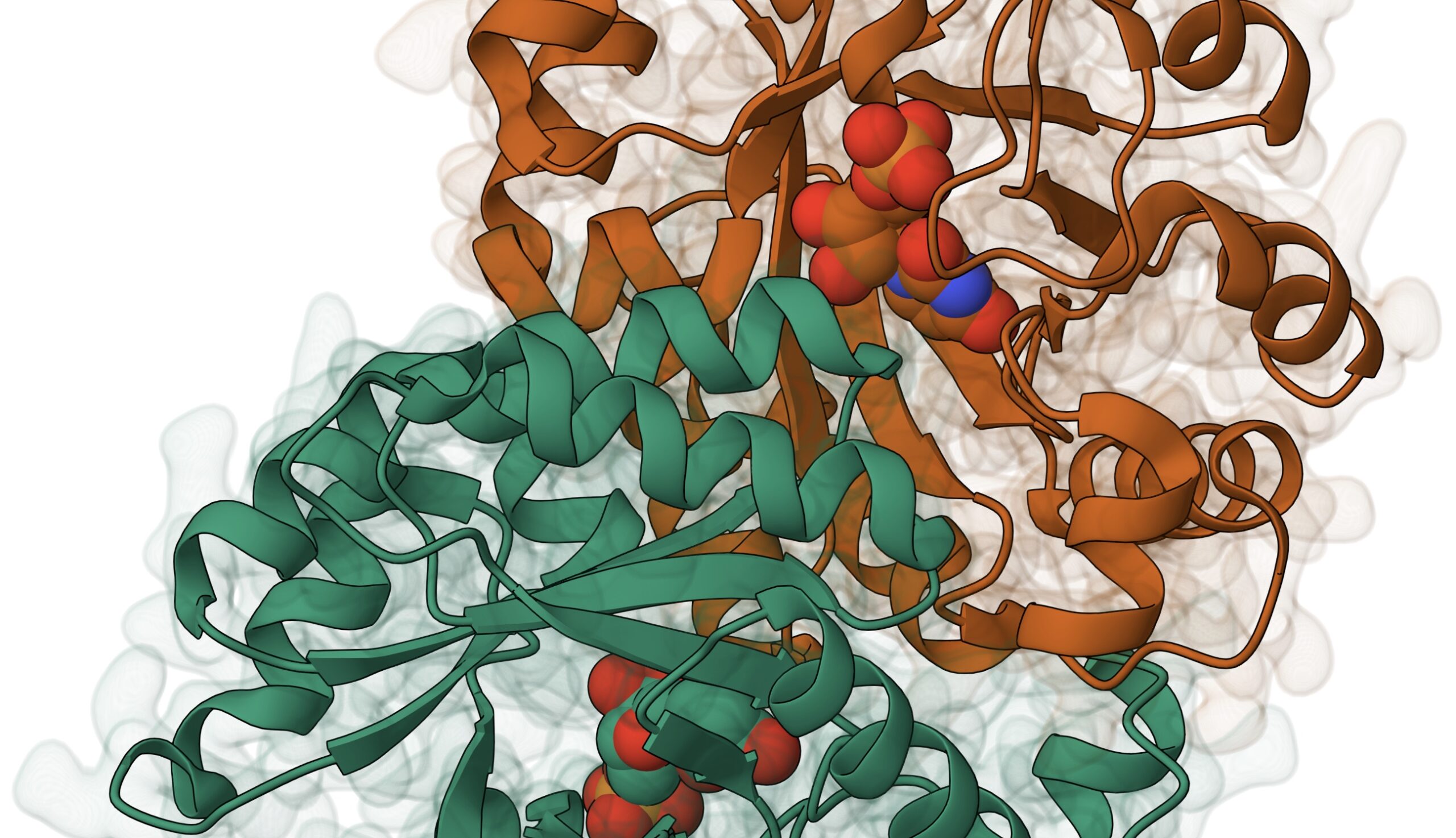
Enzymatic reactions: a peep through the centuries
It all started in the early 19th century, when a young French chemist, Anselme Payen, not yet aware he’d be etching his name in the annals of scientific history, was busily managing a borax refining facility.
He was just twenty, a time when most of us are still figuring out our path, but not Anselme. By 1820, he began collaborating with Jean-François Persoz, a pharmacist and chemist. Together, they studied barley malt.
They were onto something big, unraveling how this humble ingredient transforms starch into sugar – a process pivotal to brewing and baking. In 1833, their explorations culminated in a groundbreaking discovery – the first-ever enzyme, which they fittingly named diastase, derived from the Greek word diastasis, meaning separation. It was a natural catalyst that could break down complex starch chains into simpler, sweeter maltose.
Fast forward to the early 1900s. The biochemical nature of enzymes remained shrouded in mystery and debate, until chemist James B. Sumner’s landmark work in 1926. Just as Erwin Schrödinger was busy deriving his equation in Arosa, James B. Sumner was making his own discovery: proving that enzymes, such as urease and later catalase, are just proteins. The final nail in the debate’s coffin is hammered in by John Howard Northrop and Wendell Meredith Stanley with their research on digestive enzymes, earning them the 1946 Nobel Prize.
There we have it: enzymes are proteins. So far, so good. But what do enzymes actually do?
Further reading: A Broader View: Microbial Enzymes and Their Relevance in Industries, Medicine, and Beyond | 2013
What do enzymes actually do?
Enzymes are nature’s master catalysts. These proteins are the linchpins in a whirlwind of chemical reactions essential for, well, pretty much everything that keeps us ticking. They’re the driving force behind the smooth operation of metabolic processes in our cells, crucial for keeping the wheels of life turning.
But here’s where enzymes really steal the show: their unique three-dimensional structure.
This is what gives enzymes their incredible specificity, allowing them to pick and choose their molecular partners with remarkable precision. Enzymes excel at making reactions happen quicker by lowering the activation energy, effectively speeding up processes that could otherwise take eons – quite literally.
Take orotidine 5′-phosphate decarboxylase, for example. This enzyme catalyzes a reaction from millions of years to mere milliseconds, boasting an acceleration factor of 1017, making it the most proficient enzyme we’ve discovered so far.
Despite this remarkable specificity, enzymes can be easily influenced by their environment. Factors like pH, solvents, temperature, and inhibitors can significantly alter their activity. The latter aspect is especially important in medicine, where many drugs function by modulating these enzymatic controls.
So, here comes today’s big question: How can we simulate all of this? Can we accurately describe enzymatic reactions and estimate their activation energies with quantum chemistry?
If you’ve been following From Atoms To Words, you’ll know fully simulating an enzyme with quantum chemistry isn’t quite feasible yet. Why? Well, these systems are simply too large. Plus, we need to consider dynamics, solvent effects, temperature, entropy – the whole shebang.
But guess what? We’ve got some tricks up our sleeves – clever approximations that let us harness quantum chemistry to study enzymatic reactions.
And that brings us to the star of today’s story: the cluster approach, a highly effective method in our quantum chemistry toolkit.
Ready to dive into the nitty-gritty details?
More on From Atoms To Words:
▸ From Earth to the Cosmos: How Hydrogen Bonds Shape Life
▸ Modeling the Origins of Life: Quantum Simulations of the Primordial Soup
▸ How Can Coarse-Grained Simulations Reveal Geckos’ Wall-Clinging Skills?
Modelling enzymatic reactions with Quantum chemistry: the cluster approach
So, how do we model enzymatic reactions with quantum chemistry? Well, in the past two decades, smart quantum chemists have established and refined the so-called cluster approach. This methodology is like a high-powered microscope that zeroes in on a crucial yet small part of the enzyme around the active site, neglects the rest of the protein, and employs quantum chemistry to investigate the complexities of enzymatic reactions.
At the forefront of this approach is DFT, with the B3LYP functional emerging as a standout choice. This preference isn’t random; it’s because B3LYP finds that sweet spot between computational speed and accuracy.
Rewind about 15 to 20 years, and you’d find that a typical active-site model within the cluster approach included fewer than 50 atoms. Despite this seemingly simple makeup, these models were surprisingly adept at solving complex mechanistic problems, particularly with metalloenzymes. Fast forward to today, and the picture has dramatically changed. Thanks to rapid advancements in computing technology, active-site models now routinely boast 250 to 300 atoms. This growth speaks volumes about the cluster approach’s increasing robustness.
This method’s versatility is impressive, finding applications across a diverse range of enzyme families. Over the years, it has tackled numerous challenges, underscoring its vital role in our ongoing journey to unravel the subtle workings of enzymes.
But here’s the catch. How can we use quantum chemistry for systems of 200-300 atoms to understand enzymatic reactions, when enzymes themselves are proteins composed of hundreds of amino acids and more?
The answer lies in smart approximations. So, before diving into three illustrative case studies, let’s explore the most important approximations that make the cluster approach feasible and effective.
Further reading: Status Report on the Quantum Chemical Cluster Approach for Modeling Enzyme Reactions | 2022
The Cluster Approach: A list of smart approximations
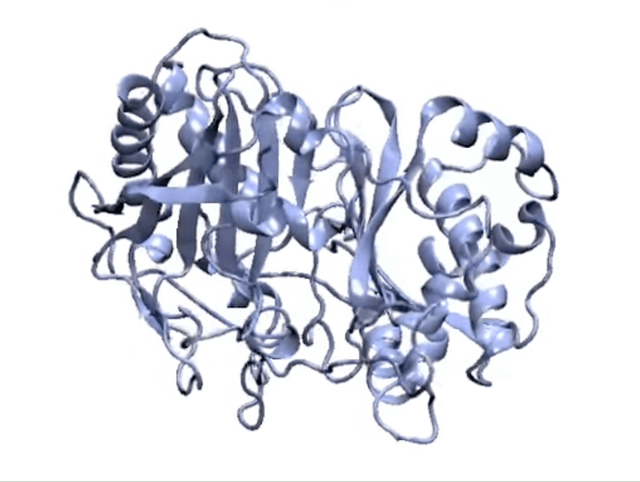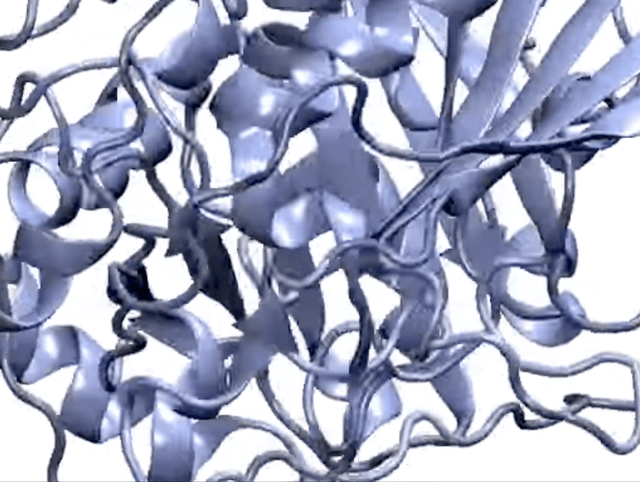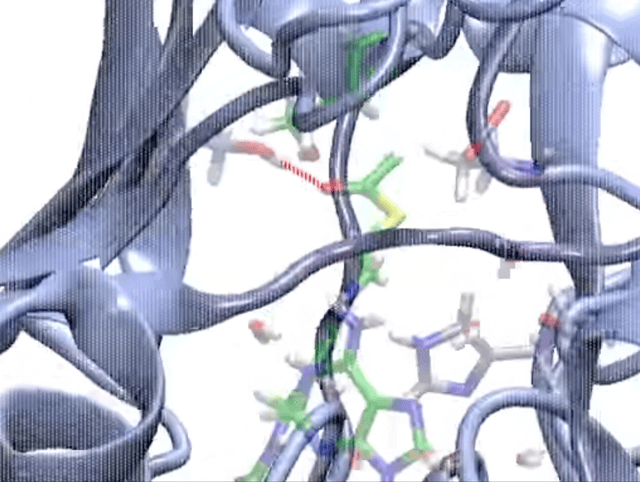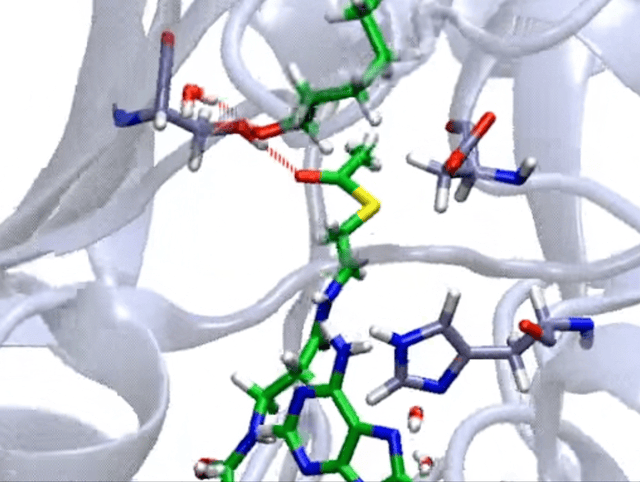
When we talk about the cluster approach, we’re focusing on a small but crucial segment of the enzyme – the active site – to dissect its properties and reaction mechanisms. It’s a clever strategy, but not without its challenges. One major hurdle is considering the influence of the rest of the enzyme, which isn’t part of our model.
So, how do we tackle this? Well, we roll up our sleeves and make some smart approximations:
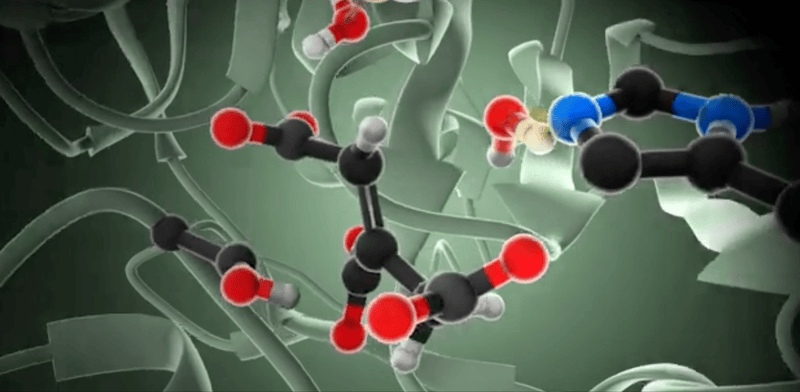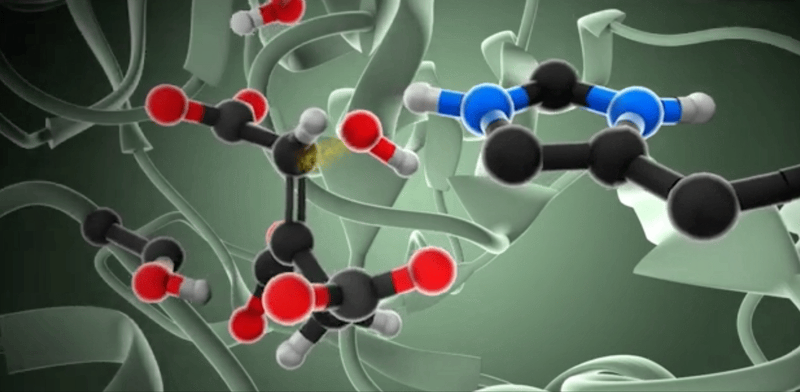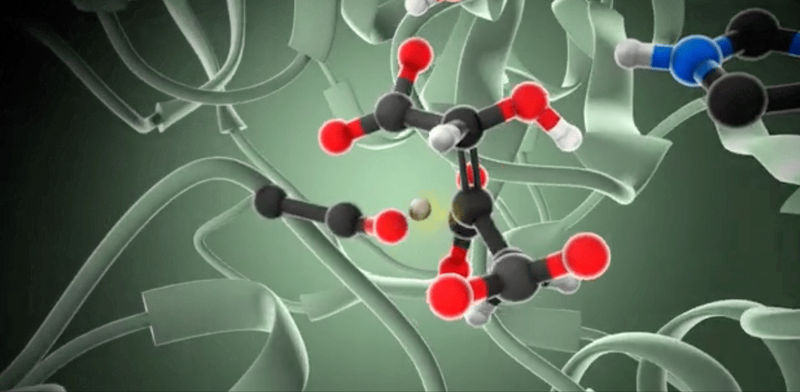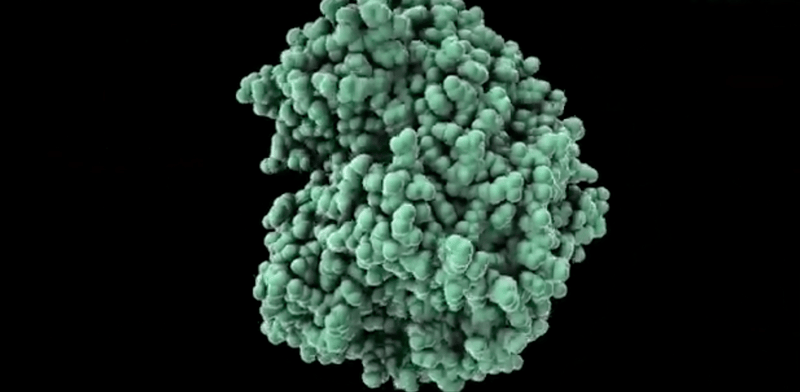
- Steric Influence Modeling: The method relies on a coordinate locking scheme. Imagine taking a snapshot of the enzyme’s active site. That’s essentially what we do here. We fix a number of atoms in their crystallographic positions, especially where our model cuts off from the rest of the enzyme structure. Starting small and gradually increasing the model size allows us to check the stability of our results, pinpoint potential errors in larger models, and get a better understanding of different parts of the enzyme.
- Implicit Solvation Models for Electrostatic Influence: The second approximation deals with the electrostatic influence of the enzyme’s surroundings. We use the so-called implicit solvation models, treating the environment around the active site of the enzyme as a homogeneous polarizable medium with a specific dielectric constant (usually set at ε = 4).
- Reaction kinetics: In applying these models, calculated energies at the quantum level are typically translated to experimental rates using classical transition state theory. It’s a generally solid approximation, although we leave out quantum tunneling effects, which are anyway minor contributors in most of the cases.
- Entropic Effects: A crucial aspect of the cluster approach is its focus on the chemical step of enzymatic reactions, starting from the enzyme-substrate complex. Since entropy changes during these steps are usually small, we simplify things by approximating free energy with enthalpy. This means that with the cluster approach we usually neglect entropic changes related to substrate binding and product releasing.
Despite these approximations, the cluster approach remains a solid method in enzymatic research. It allows us to swiftly evaluate various reaction mechanisms through detailed energy calculations. So, are you ready to see this approach in action? Brace yourself for a technical deep dive.
Further reading: Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions | 2017
More on From Atoms To Words:
▸ The Evolution of Quantum Chemistry: From Pencil and Paper to Quantum Computing
▸ Computational Chemistry 2043: A Quantum Peep into the Future
▸ Multiscale Simulations of DNA: From Quantum Effects To Mesoscopic Processes
Quantum chemistry of enzymatic reactions: three case studies
1. Investigating Biochemical Processes
- Study: Peptide Release on the Ribosome Involves Substrate-Assisted Base Catalysis, 2016
- Enzymatic System: This study focused on the peptidyl-tRNA hydrolysis mechanism in the ribosome, a critical process in the termination of protein synthesis.
- Purpose: A cluster model with 224 atoms was employed to investigate the peptidyl-tRNA hydrolysis reaction mechanism at the ribosome’s peptidyl transferase center. This mechanism involves the hydrolysis of an ester bond between the P-site tRNA and the nascent peptide chain, a reaction known to be pH-dependent. The aim was to understand the specific steps and energy barriers involved in this essential biochemical process.
- Key Learning: The main takeaway was the identification of the reaction initiation step: the deprotonation of the P-site A76 2′-OH group, which activates the water nucleophile. The study accurately estimated the energy barriers for subsequent steps, revealing an overall activation energy consistent with experimental kinetics. This finding aligns with the pH-dependence observed in experiments, highlighting the cluster approach’s strength in elucidating complex enzymatic mechanisms.
2. Understanding Enzymatic Selectivity
- Study: Theoretical Study of the Reaction Mechanism of Phenolic Acid Decarboxylase, 2015
- Enzymatic System: Phenolic acid decarboxylase catalyzes the nonoxidative decarboxylation of phenolic acids into p-vinyl derivatives without cofactors. This enzyme is of significant biocatalytic interest, especially for applications in polymer and food industries.
- Purpose: A cluster model with over 300 atoms was designed to explore different mechanistic scenarios for phenolic acid decarboxylase. The model aimed to clarify the role of specific residues in the reaction and to understand the enzyme’s activities, such as carboxylation and hydration of hydroxystyrenes.
- Key Learning: While correcting previous hypotheses about the orientation of substrate binding, the study revealed that Glu64 acts as a general acid in the reaction. Calculations showed that the overall energy barriers for key steps in the reaction are consistent with measured rate constants. The model successfully replicated the enantioselectivity observed in the hydration reaction, offering insights into the mechanism’s subtleties and identifying critical active-site residues. This underscores the cluster approach’s ability to provide detailed, accurate predictions of enzymatic activities and selectivities.
3. Modeling Enantioselectivity
- Study: Quantum Chemistry as a Tool in Asymmetric Biocatalysis: Limonene Epoxide Hydrolase Test Case, 2013
- Enzymatic System: The focus was on the stereoselectivity of enzymes, particularly the enantioselectivity of limonene epoxide hydrolase, an enzyme that catalyzes the hydrolysis of epoxides into vicinal diols. This study is significant for its implications in asymmetric biocatalysis, a field crucial for producing specific chiral molecules used in various industries.
- Purpose: The cluster approach was employed to understand and reproduce the enantioselectivity observed in enzymes. This involved creating active-site models of about 250 atoms to accurately account for the chiral environment around the substrate and to discern the effects influencing selectivity. The goal was to elucidate the sources of enantioselectivity in limonene epoxide hydrolase and other enzymes by examining the regioselectivity of ring openings in various substrates.
- Key Learning: The study successfully matched experimental findings, especially in terms of the small energy differences crucial for determining selectivity. It provided a detailed understanding of how different mutations in limonene epoxide hydrolase affect enantioselectivity, offering a rational explanation for the observed variations. This example highlights the cluster approach’s effectiveness in accurately modeling and predicting the intricate details of enzyme selectivity, cementing its value in advancing the field of asymmetric biocatalysis.
More on From Atoms To Words:
▸ 60 Years in the Making: AlphaFold’s Historical Breakthrough in Protein Structure Prediction
▸ AI in Drug Discovery: Chasing Dreams, Facing Realities
▸ Is Machine Learning Going to Replace Computational Chemists?
A final personal touch
In today’s story, we’ve journeyed through the applications of the cluster approach in studying enzymatic reactions, a method that’s really held its own for the past two decades.
Now, let’s be real: the cluster approach is far from being perfect, especially when you consider all the approximations involved. And there are more sophisticated approaches at hand, think of hybrid quantum mechanical/molecular mechanical (QM/MM) schemes. But, the cluster approach does offer a pretty solid peek into how enzymatic reactions work and their energetics.
The case studies we looked at really show off what this approach can do. It’s spot-on in pinpointing reaction steps, shedding light on how enzymes interact with substrates, and at predicting enantioselectivity, all while offering a deeper understanding of complex enzymatic reactions.
What I love about it, is how it slices through the complexity of biochemical mechanisms, giving us an atomistic view of enzyme behavior and reactivity. For me, the cluster approach is a pretty nifty tool in the computational chemist’s toolkit. It definitely was for my research on photosystem II.
So, dive in, play with it, have fun.
If you enjoyed this dive into quantum chemistry and enzymatic reactions, I’d love to hear your thoughts. Agree, disagree, or have a totally wild theory of your own? Let’s connect! Subscribe to my LinkedIn newsletter and let’s keep the conversation rolling.