The first time I used all-atom molecular dynamics, I, a quantum chemist, was completely in awe. Imagine thousands of atoms, moving around in what seems like chaos, eventually forming a structure that matches what we see in experiments. This was right after my PhD, when I returned to Italy for a couple of years to do my first post-doc at the International School for Advanced Studies in the stunning city of Trieste. Oh, how I adore that place! I remember those lunch breaks when I’d jump on my motorbike and head out for a quick swim. Then, returning to the lab, I’d be greeted by those amazing all-atom molecular dynamics simulations of DNA fragments. Years later, in Sardinia, I was still immersed in the blue sea and in molecular dynamics, this time of respiratory proteins. The whole simulation process was a painstaking affair; on a supercomputer, it would take weeks to reach significant dynamics of about tens of nanoseconds. That was almost two decades ago. And today? Now, we’re at the forefront of molecular dynamics, running simulations on a colossal scale. The progress is astonishing. Curious to see how far we’ve really come? Buckle up then, because today we’re diving into all-atom molecular dynamics of SARS-CoV-2, exploring simulations of 305 million atoms. Let’s go.

The Computational Microscope of the Biological Underworld
I am talking about all-atom molecular dynamics. Which is like a detailed movie of the microscopic world that portrays every single atom in the studied system. And just like prepping for a movie production, starting a molecular dynamics simulation requires some groundwork. First and most importantly, you need a force-field—a set of parameters usually determined by ab initio methods, in other words: more rigorous and quantum-based.
See, unlike ab initio methods, such as quantum chemistry’s Hartree-Fock and beyond, where you define a structural input and let the laws of quantum mechanics work their magic, molecular dynamics is in a league of its own.
You assign each atom, whether it’s carbon, oxygen, or any element really, a unique set of parameters that reflect their specific type. So, for each type, the parameters vary depending on whether the atom is, say, in a double bond, nestled in an aromatic ring, or part of an aliphatic chain. Imagine every atom type, each with its specific set of parameters – charge, van der Waals, hydrogen-bonding parameters, etc. – all coming together in a grand performance that unfolds according to the laws of classical physics.
And guess what? It works!
Giant molecules like titin, which holds the current world record for the largest protein, have been brought to life through all-atom molecular dynamics simulations. And boy, have these simulations demonstrated extraordinary capabilities.

But molecular dynamics isn’t limited to biomolecules; it’s a versatile tool across various scientific domains. At Quantistry, for example, we apply it to the investigation of systems like alloys, polymers, electrolytes, with the aim of rationalizing, optimizing, and designing. I myself played with heme-proteins during my academic career, like hemoglobin and myoglobin to explore the journey of a little molecular ligand from the inside of the protein to the outside. Now, here’s where the power of molecular dynamics really shines: its ability to start-off from experimental structural information and extend to its dynamical behavior, thus providing juicy scientific insights.
But let’s be clear – all-atom molecular dynamics isn’t without its quirks and quibbles. The computational gymnastics required to create these simulations, along with the challenges of capturing larger biomolecules over longer timescales, have been significant. And then there are the questions about the accuracy of these simulations, especially given the known approximations in the force fields used.
To leap over these hurdles, researchers have been upping their game, scaling up all-atom molecular dynamics simulations to tackle megasystems, at least from the point of view of a humble quantum chemist like me, such as ribosomes and entire viral capsids. Or to simulating smaller systems for durations stretching to milliseconds and beyond.
The spatial and time advancement in molecular dynamics, especially for biologically relevant systems, is what led to the concept of the computational microscope. It’s a term coined by Lee in 2009 that encapsulates the essence of all-atom molecular dynamics simulations, a technique that’s become the go-to tool for exploring the function and dynamics of the biological underworld.
So, today, I want to take you through an impressive example of computational microscopy – the simulations by Casalino et al. of the SARS-CoV-2 virus. Get ready to be amazed.
Further reading:
▸ Discovery Through the Computational Microscope | Structure, 2009
▸ The world’s largest proteins? These mega-molecules turn bacteria into predators | Nature, 2023

More on From Atoms To Words:
▸ Computational Chemistry 2043: A Quantum Peep into the Future
▸ The Evolution of Quantum Chemistry: From Pencil and Paper to Quantum Computing
▸ Digital Alchemy: Computers in Chemistry and the Future of Scientific Discovery
All-Atom Molecular Dynamics of SARS-CoV-2
Alright, let’s dive into the nitty-gritty of this COVID-19 virus, but let’s skip the grim statistics – we’ve all been bombarded enough with those. We’re here to unravel the mysteries of the virus itself, SARS-CoV-2, the little bugger that’s been causing all that mayhem.
You see, scientists around the globe have been on a mission: to crack the code of this virus, to suss out a better vaccine or a better cure, and to really get a grip on how the virus ticks – its transmission, its infectivity, the whole shebang.
Now, if we zoom in a bit, we start to see that understanding the virus structure and dynamics is a bit like trying to solve a puzzle with half the pieces missing.
Traditionally, we’ve got our experimental heavy hitters like X-ray crystallography, cryo-EM, and cryo-ET. They give us these incredible structural snapshots of the virus proteins, showing us photographs of how they interact with the host cells.
Pulling all the data together into a clear picture? That’s been a bit of a roadblock towards obtaining a unified model that can comprehensively explain the virus infectivity.
Plus, let’s not forget that these high-resolution structural data have their blind spots. We’re talking about the dynamic, the wiggly atoms of the flexible parts of the proteins and those sugar-like groups on the virus surface – the glycans. These elusive characters are crucial to the virus life cycle, yet they’re often just out of reach in our static snapshots.
Welcome team Casalino! They’ve cooked up this innovative, AI-driven workflow that’s like a supercomputer on steroids. It crunches the numbers, parallelizing all-atom molecular dynamics simulations to explore the inner workings of the SARS-CoV-2 virus.
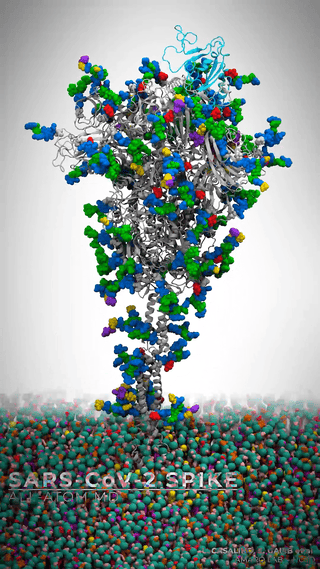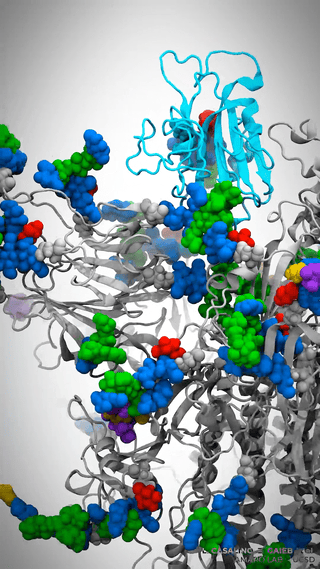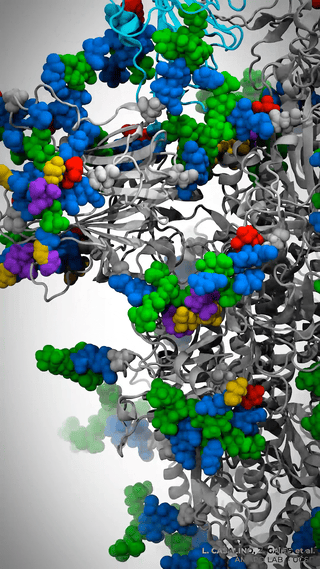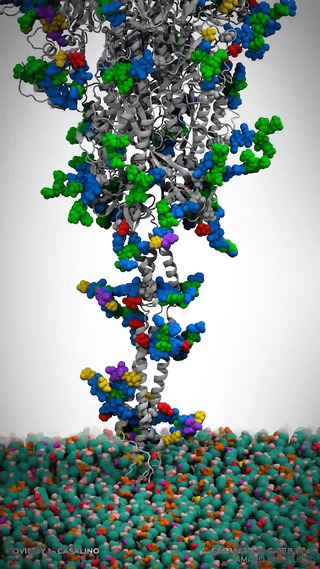
This is where it gets really cool – Casalino et al. aren’t just looking at the dynamics of the spike protein across various environments; they’re simulating the whole viral envelope. We’re talking about a model of a whopping 305 million atoms.
Man, let’s pause here for a sec. Let that sink in! 305 million atoms. My poor quantum chemist brain gets all on fire trying to process that amount of information.
What about you? Wanna give it a try? Let’s dive deeper then.
Further reading: AI-driven multiscale simulations illuminate mechanisms of SARS-CoV-2 spike dynamics, 2021
More on From Atoms To Words:
▸ ReaxFF Molecular Dynamics: Simulating Complexity Beyond Quantum Chemistry
▸ Bridging Theory and Experiment: 14 Reasons Chemical Simulations Stand as the Third Pillar of R&D
▸ When Will RNA Structure Prediction Get Its AlphaFold Breakthrough?
Casalino’s All-Atom Molecular Dynamics of SARS-CoV-2: How To?
So, what did Casalino et al. really pull off in their study? Well, hold onto your hats because this is where it gets both brainy and cool.
In a nutshell: they meticulously gathered different pieces of experimental structural information of the SARS-CoV-2 virus, mounted together, filled the missing pieces and crafted a comprehensive model of it. Then, they brought this model to life, with all-atom molecular dynamics simulations on space- and time-scales hardly seen before.
This approach gives us insights that are usually tricky if not impossible to get experimentally. We’re talking about the dynamical features of the virus mechanism of action, the nitty-gritty details that are key to understanding the whole infection process. And why does this matter? Because it’s a crucial step towards figuring out potential therapies or improving the ones we have.
Now, to really get a grasp of the scale of their achievement, let’s dive into the technicalities. Here’s a quick overview of Casalino’s models and simulations:
- Spike Protein Models:
- Model Size: Two all-atom models, each with 1.7 million atoms. They were built starting from the cryo-EM structures of the spike in the open state.
- Time Scale: Simulations of ∼4.2 µs and ∼1.7 µs for open and closed states.
- Purpose: To study the structural dynamics crucial for the virus entry into human cells.
- Simulations of the Receptor Binding Domain-ACE2 Complex:
- Model Size: Each simulation involved approximately 800,000 atoms.
- Time Scale: Extensive sampling to examine the interactions. Similar time scales as in the Spike Protein Models.
- Purpose: To gain insights into the interaction between the virus and human cell receptors.
- Spike Opening Simulations:
- Model Size: Large-scale simulations using the weighted ensemble method.
- Time Scale: Sampling of ∼7.5 µs in initial runs, and ∼70.0 µs in extended simulations.
- Purpose: To observe the transition of the spike protein from closed to open states (activation and binding to ACE2 receptors).
- Two-Parallel-Membrane System of the Spike-ACE2 Complex:
- Model Size: All-atom complex with over 8.5 million atoms.
- Time Scale: Overall simulation time of ∼702 ns.
- Purpose: To understand the virus entry mechanism through the spike-ACE2 interaction.
- Full-Scale SARS-CoV-2 Viral Envelope Simulation:
- Model Size: A system containing about 305 million atoms.
- Time Scale: Production simulations running for 84 ns.
- Purpose: To replicate the entire viral envelope, enabling study under realistic biological conditions.
How long did it take to run these simulations? Between 8 and 26 days. Impressive, no doubt. But here’s the big question: Is this just a computational exercise, or can we glean meaningful scientific insights from such astounding simulations?
Further reading: AI-driven multiscale simulations illuminate mechanisms of SARS-CoV-2 spike dynamics, 2021

More on From Atoms To Words:
▸ Multiscale Simulations of DNA: From Quantum Effects To Mesoscopic Processes
▸ From Earth to the Cosmos: How Hydrogen Bonds Shape Life
▸ AI in Drug Discovery: Chasing Dreams, Facing Realities
All-Atom Molecular Dynamics of SARS-CoV-2: What do we learn?
Computationally speaking, these all-atom molecular dynamics simulations are phenomenal. But it’s more than that: Casalino and team have handed us a high-definition, atomic-level, dynamical map of the SARS-CoV-2 virus. With their sophisticated models, they’ve peeled back layers of the virus, revealing its structure and dynamics in unprecedented detail. This is the kind of insight that could fill knowledge gaps in virology and molecular biology.
So, what scientific insights do we learn from Casalino’s deep dive into all-atom molecular dynamics of the SARS-CoV-2 virus?
- Glycan Shield Characterization
The study in-depth analysis of the spike protein glycan shield, including its stalk, is a major advancement. The discovery of the roles of N-glycans at N165 and N234 in modulating the spike’s receptor-binding domain dynamics improves our understanding of the virus infection mechanism. - Spike Protein and ACE2 Receptor Interaction
The identification of a flexible hinge in the human ACE2 receptor, along with the spike protein flexibility in ACE2 binding, provides key insights for therapeutic development. This flexibility is likely crucial in forming complexes with the spike protein, essential for cell fusion and viral entry, suggesting new strategies for disrupting the virus ability to infect human cells and guiding vaccine and drug development. - Unbiased Pathways of Spike Receptor Binding Domain Transition
Casalino’s mapping of the unbiased pathways of the spike’s receptor binding domain transition from closed to open states illuminates paths for drug discovery. This understanding is vital for identifying novel druggable pockets within the spike protein, leading to the development of more effective COVID-19 treatments. - SARS-CoV-2 Viral Envelope Simulation
Simulating the SARS-CoV-2 viral envelope with 305 million atoms represents a significant leap in virological research. This all-atom molecular dynamics simulation sets a new benchmark for studying viruses under more realistic biological conditions, enhancing our understanding of their behavior and infection mechanisms. - AI-Driven Adaptive MD and Workflow for Multiscale Simulation
The integration of AI in molecular dynamics approaches and the development of a scalable workflow for multiscale simulations represent technological milestones. This study not only enhances the scope of exploring the spike protein conformational space but also broadens the implications for other molecular systems, accelerating the all-atom modeling of complex viral mechanisms and extending its applications to giant biological structures.
Further reading: AI-driven multiscale simulations illuminate mechanisms of SARS-CoV-2 spike dynamics, 2021
More on From Atoms To Words:
▸ How Can Coarse-Grained Simulations Reveal Geckos’ Wall-Clinging Skills?
▸ 60 Years in the Making: AlphaFold’s Historical Breakthrough in Protein Structure Prediction
▸ Is Machine Learning Going to Replace Computational Chemists?
A Final personal touch
What I love about Casalino and team’s work is the way they’re stretching the limits of all-atom molecular dynamics. Their stunning simulations of the SARS-CoV-2 virus aren’t just a one-off marvel; it’s a sneak peek into a future where such detailed simulations become the norm.
Imagine the possibilities!
This opens up an exhilarating path for therapeutic development. We’re talking about a future where simulating complex systems with all-atom molecular dynamics becomes part of the routine toolkit. And when you think about combining this with the leaps being made in machine learning for protein structure prediction and drug discovery, it’s clear we’re on the cusp of a new era.
This is about merging computational ingenuity with experimental R&D in ways we’ve only just begun to explore.
And that, folks, is something to be truly excited about.
If you enjoyed this dive into computational microscopy and all-atom molecular dynamics simulations, I’d love to hear your thoughts. Agree, disagree, or have a totally wild theory of your own? Let’s connect! Subscribe to my LinkedIn newsletter and let’s keep the conversation rolling.