It had only been a few days since the world shook with news of a scientific triumph in protein structure prediction. Word had spread about an insurmountable biological puzzle that had tormented scientists for decades, and lo and behold, they cracked it. The hot topic on everyone’s lips was protein structure prediction – the ability to decipher how a protein folds and behaves solely from its amino-acid sequence. I must confess, I played the skeptic initially, but then this discovery grabbed everybody’s attention. Including mine. And you know who stole the spotlight? Machine learning. Today, we are going to revisit the breakthrough in protein structure prediction achieved by the mighty AlphaFold. But before we dive into the solution, let us first appreciate the mission impossible it set out to conquer.

Why is protein structure prediction such a mission impossible?
Cast your mind back to the 1960s, a time of great scientific fervor following the discovery of the DNA double helix. There was a hopeful anticipation that protein structures would also exhibit some elegant internal pattern and regularity. Linus Pauling had even foreseen the existence of α-helices. But the unveiling of myoglobin’s structure threw a wrench in those assumptions, presenting helices haphazardly packed together. This marked the starting point of the vexing protein folding problem.
So, what makes predicting how a protein folds from its amino-acid sequence such an enormous challenge?
The thing is, as pointed out in 1969 by Cyrus Levinthal, an unfolded protein possesses an astronomical number of potential configurations. Literally.
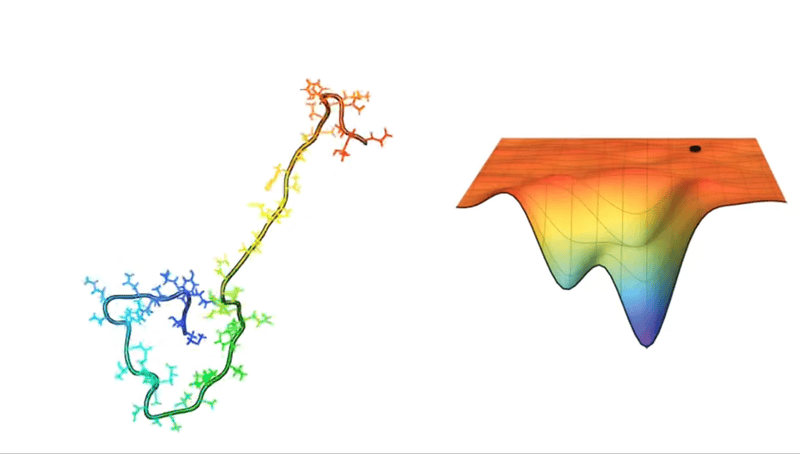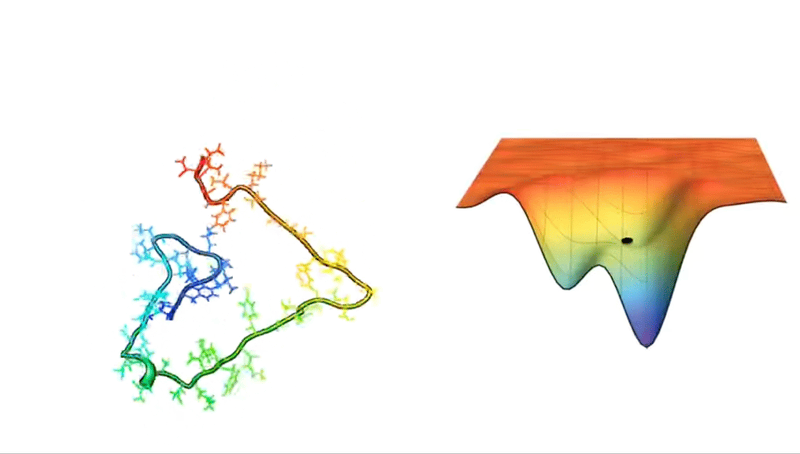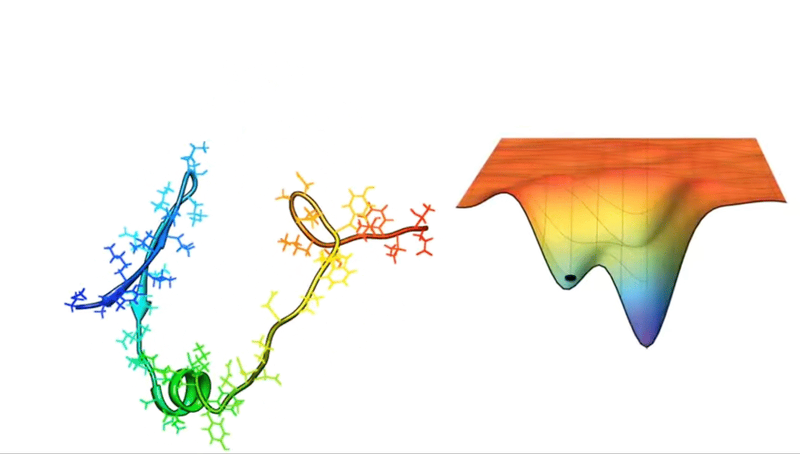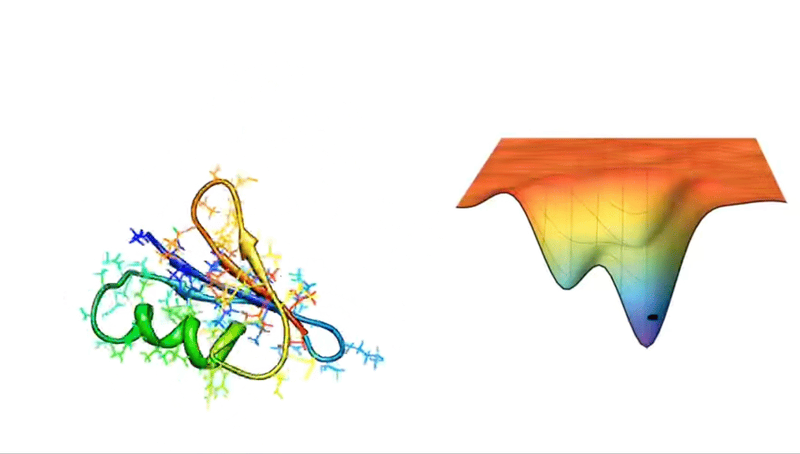
For example, consider a protein chain with 100 residues. This has 99 peptide bonds, which result in 198 distinct phi and psi bond angles (see Figure above). Now, imagine that each of these angles can take on one of three possible conformations. This amounts to 3198 potential conformations! And if the protein were to explore all of them, the folding would take longer than the age of the universe.
Astonishingly, most proteins manage to fold into their correct structures within milliseconds or microseconds—such a mind-boggling conundrum has even been given a fancy name: Levinthal’s paradox.
Faced with such hellish combinatorics, early attempts to crack the protein structure puzzle relied on databases and secondary structure prediction algorithms or computational methods, such as force fields and Monte Carlo sampling. It was a valiant effort, one that did propel some progress in the mid-1980s.
Then in 1994, a maverick by the name of John Moult stepped onto the scene with his brainchild: the Critical Assessment of Techniques for Protein Structure Prediction, or CASP for short. This is a biennial competition where research groups are invited to take part in a blind test. Their mission? To predict protein structures solely from the amino-acid sequence, without any knowledge of the experimental results.
Over the course of grueling decades, advancement moved forward inch by painful inch until the glorious arrival of AlphaFold in 2020. This game-changer harnessed the untamed power of machine learning and pointed it directly at the problem.
AlphaFold’s results left everybody with their jaws on the floor. More than 60 years since the first experimental protein structures were discovered, its performance outperformed all reasonable forecasts. The reverberations of this scientific triumph extended far beyond the confines of its immediate domain, shaking the very foundations of what we thought was scientifically possible.
For more on the protein structure prediction conundrum, check this, this, and this.
More on From Atoms To Words:
▸ Is Machine Learning Going to Replace Computational Chemists?
▸ AI in Drug Discovery: Chasing Dreams, Facing Realities
▸ Chemical Space to Material Discovery: Simulations and Machine Learning Leading the Way
AlphaFold: Breaking the barrier of Protein Structure Prediction
The air was thick with anticipation in the lead-up to the CASP14 meeting, as the unveiling of the long-awaited results ignited a storm of excitement that spread like wildfire across the Twitterverse. A collective gasp echoed throughout the audience, as they fixated their gaze on the screens, spellbound by a bar plot that defied every expectation. Among the competitor research teams, one emerged as an absolute victor: Group 427, the at-the-time enigmatic AlphaFold2.

The true power of AlphaFold lies within the numbers. So, let’s talk root-mean-square deviation, or RMSD. If you are not familiar with it, just remember this: a lower RMSD indicates a prediction of superior quality, as it signifies a smaller deviation from an experimental structure.
For the top 95% of predicted amino acids, AlphaFold’s results showed an RMSD of a mere 0.96 Å when compared to experimental models. In stark contrast, the next-best method lagged far behind at 2.83 Å.
What does it mean?
AlphaFold consistently reproduced experimentally determined structures with jaw-dropping accuracy.
After decades of fierce competition, the organizers of CASP14 made a proclamation of historic significance. AlphaFold, this marvel of machine learning, had achieved what had eluded the scientific community for a staggering 60 years: the ability to predict a protein’s structure solely based on its sequence.
The holy grail had finally been unearthed.
How good is AlphaFold, exactly? Astoundingly so.
Oxford Protein Informatics Group
How does AlphaFold do its Protein Structure Prediction magic?
At the heart of AlphaFold‘s triumph lies a wealth of data sourced from two significant channels. The first is the Protein Data Bank, a global repository housing an impressive collection of over 180,000 experimental protein structures. These have been meticulously cataloged by the dedicated structural biology community for several decades, serving as a beacon of knowledge since the 1970s.
The second wellspring of information originates from the realm of protein sequences. This expansive domain owes its existence to the unwavering efforts of scientists worldwide, who have contributed their expertise to compile data through ambitious genome sequencing initiatives. Thanks to these publicly accessible resources, AlphaFold can scrutinize the connections between established protein structures and the vast array of protein sequences.
From this unique perspective, AlphaFold’s advanced algorithms spring to life, deploying their remarkable capabilities to generate accurate predictions of protein structures. But that’s not all—it fearlessly ventures into uncharted territories, providing precise predictions for amino-acid sequences it has never encountered before.
AlphaFold’s extraordinary capabilities are a result of ingenious solutions and the synergistic application of cutting-edge techniques. Deep learning algorithms are harnessed to leverage the insights derived from decades of experimental data. Multi-sequence analysis plays a pivotal role in AlphaFold’s engine, leveraging conserved peptide structures and evolutionarily coupled residues to unravel the complexities of protein folding.
AlphaFold’s accomplishments weren’t a stroke of luck. They were made possible by a combination of innovative techniques, colossal processing power, and extensive computational time. Through a harmonious blend of human ingenuity and the brute force of machine learning, AlphaFold cracked the code for how an amino-acid sequence folds into the expanse of three-dimensional space.
For more nitty-gritty technical details, check out the awesome blog post by the Oxford Protein Informatics Group titled: “CASP14: what Google DeepMind’s AlphaFold 2 really achieved, and what it means for protein folding, biology and bioinformatics.”

More on From Atoms To Words:
▸ All-Atom Molecular Dynamics of SARS-CoV-2: The Computational Microscope’s View of 305 Million Atoms
▸ Large Language Models for Chemistry: Is the Beginning of a New Era?
▸ 7 Noncovalent Interactions in Proteins: The Hidden Architects of Structures and Functions
Even AlphaFold has some limitations
By deciphering the enigma of protein folding, AlphaFold has opened up new horizons in biology, bioinformatics, and our understanding of life itself. But hey, let’s keep it real, AlphaFold ain’t perfect. There are some limitations we need to be aware of.
Here’s the deal.
You see, many proteins like to team up and form complexes with other proteins, nucleic acids (DNA or RNA), or ligands. AlphaFold struggles to predict 3D structures of this kind.
Another point to consider: proteins are dynamic beasts. They can switch up their structures depending on their surroundings or where they’re at in their functional cycle. However, AlphaFold usually sticks to producing just one conformation. This leaves us with a bunch of questions about the dynamics of the predicted proteins and how this relates to their biological function.
Now, when it comes to those regions of proteins that are naturally disorderly or unstructured, AlphaFold lacks the confidence to make accurate predictions. The structures it spits out for these regions might look all stretched out and ribbon-like. Don’t get me wrong, AlphaFold can be handy for identifying these regions, but it doesn’t give us any insight into the likelihood of different conformations.
And here’s some more: AlphaFold hasn’t been specifically trained or validated to predict how mutations can mess things up. So, don’t expect it to capture the effects of point mutations that can destabilize a protein.
There you have it: AlphaFold is an absolutely exciting tool. But it’s got its boundaries. Don’t worry though, the field of protein structure prediction has never been so alive, and I’m sure the brilliant minds behind AlphaFold and other machine-learning tools will continue to push the envelope and finding ways to tackle these limitations.
For more on AlphaFold limitations, check out this article published on EMBL Communications.

What does it all mean for the future of research?
This groundbreaking advancement in protein structure prediction has sparked a tremendous wave of enthusiasm within the scientific community, and with good reason.
Above all, AlphaFold is poised to revolutionize structural biology research, allowing scientists to delve into the complexities of proteins like never before.
But that’s not the end of the story. AlphaFold opens doors for investigating biological systems where high-resolution experimental data are lacking. With the invaluable support of AlphaFold, we can now formulate educated hypotheses about the inner workings of these systems.
And here’s the most thrilling aspect: the impact on drug discovery. AlphaFold’s models are set to revolutionize this field as well, significantly advancing our understanding of how drugs interact with proteins and facilitating the identification of potential targets.
More on From Atoms To Words:
▸ When Will RNA Structure Prediction Get Its AlphaFold Breakthrough?
▸ Digital Alchemy: Computers in Chemistry and the Future of Scientific Discovery
▸ Water’s Hydrogen Bonds: What Makes Them Vital for Life As We Know It?
A final personal touch
Man, when I think about the astonishing success of AlphaFold, it takes me back to those pivotal moments in scientific history that made us question everything. You know, like when we figured out that the universe’s expansion actually accelerating or when we learned that spacetime is bending under the weight of massive objects.
And now, here we are, with AlphaFold causing a seismic shift. Biology will never be the same again.
So, get ready my friends, for the future of biology is shining brighter than ever, all thanks to the monumental progress of computers and machine learning. We’re standing on the edge of a new era of discovery and as Carl Sagan would say, somewhere, something incredible is waiting to be known.
If you enjoyed this dive into protein structure prediction, I’d love to hear your thoughts. Agree, disagree, or have a totally wild theory of your own? Let’s connect! Subscribe to my LinkedIn newsletter and let’s keep the conversation rolling.