One of the things I’ve always loved about quantum chemistry is studying chemical reactions. I mean, first off: how amazing are chemical reactions? You mix together substances that seem mundane and boring, and suddenly, they transform into fairy-tale crystal trees sprouting before your very eyes. And then there’s the second marvel: simulating those reactions. I remember the excitement when I started my PhD, working on the hydrolysis of the anticancer agent, cisplatin. The activation energies I calculated on my computer were virtually indistinguishable from those measured in real-world experiments. How incredible is that? For me, it was proof that quantum mechanics really works. But you know what they say: to a quantum chemist, every science problem looks like a quantum chemistry problem. Unfortunately, that’s not always the case. I’ve come to believe that the best approach to address your experimental challenges is to use whatever computational tools are necessary. Sometimes, you have to let go of electrons and embrace multiscale approaches, like molecular dynamics or even coarse-graining. Is there a middle ground? Can we model larger systems, include environmental effects explicitly, and still capture reactivity? The answer is yes, with ReaxFF molecular dynamics. And that, dear reader, is what we’re diving into today. Are you ready? Let’s go.

Beyond quantum chemistry: Reactive molecular dynamics
Let’s say you want to explore reactivity in complex systems—be it within a battery cell, an enzyme, or nucleobases in RNA.
Where do you start?
Quantum chemistry may be your first thought, with Density Functional Theory (our friend DFT) leading the charge. It’s a logical choice that offers the potential for not only accuracy but predictive insights as well.
But to study a system with thousands or more atoms at a quantum chemistry level means one thing: approximations.
We’ve touched on this on From Atoms to Words, when we discussed the cluster approach for enzymatic reactions and the hybrid quantum mechanics/molecular mechanics method for surface adsorption. But what happens when these approximations fall short, especially when the system’s dynamics, or the impact of solvents and the environment, play a pivotal role?
You might lean towards ab initio molecular dynamics, a technique we’ve also encountered when we talked about multiscale simulations of DNA. However, this path, too, is fraught with limitations—computational, to be precise.
There is no doubt. Quantum chemistry’s allure has grown, thanks to the proliferation of user-friendly software that has made sophisticated quantum mechanical calculations more accessible. This has been a boon across various fields, from drug development to materials science, where insights from quantum chemistry guide researchers towards promising solutions while helping to sift through less viable options.
Yet, the brilliance of quantum chemistry casts long shadows. The very atomistic insight that makes quantum chemistry invaluable also demands a steep computational toll, constraining simulations to smaller scales and shorter timeframes.
This limitation is more than a mere technicality; it represents a significant bottleneck in our quest to understand and predict the behavior of complex systems.
So, what’s next?
You might consider turning to all-atom molecular dynamics, a topic we’ve covered extensively, from the dynamics of proteins and DNA to modeling viruses. Although all-atom molecular dynamics is undeniably powerful, for the chemist keen on capturing chemical reactivity, there are significant limitations. Is the cytosine in your DNA oxidizing? Is your amino acid being protonated? Is your electrolyte degrading?
Well, the force fields—the parameters used in classical molecular dynamics—lack the capability to capture such chemical reactivity. Classical molecular dynamics maintains the initial structure throughout; it can model dynamics and non-covalent interactions but doesn’t quite cut it when it comes to breaking/forming covalent bonds.
So, shall we turn off our supercomputer in frustration? Of course not.
Introducing reactive force fields, or ReaxFF for short—a beacon of hope for chemists and material scientists alike.
But what exactly is ReaxFF molecular dynamics? Let’s have a deeper look.
[Of course, another way is the machine learning way. Check it out!]
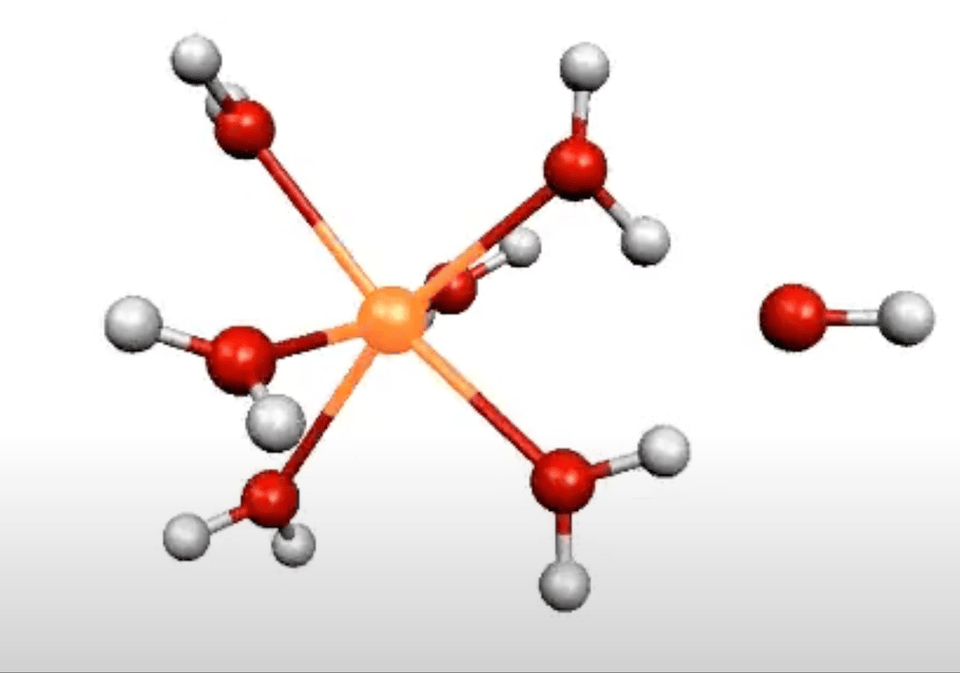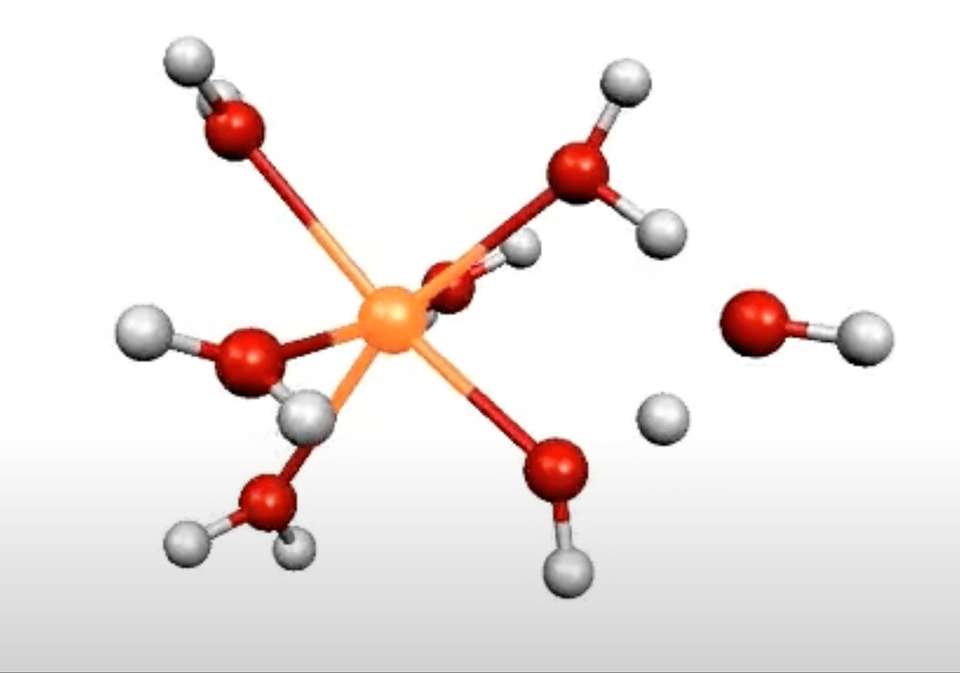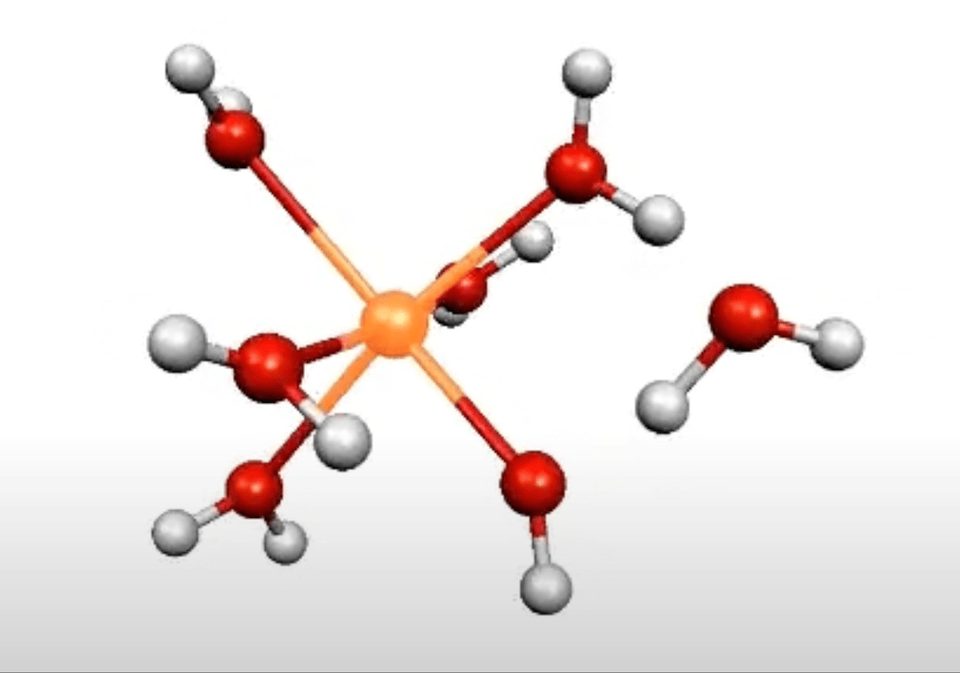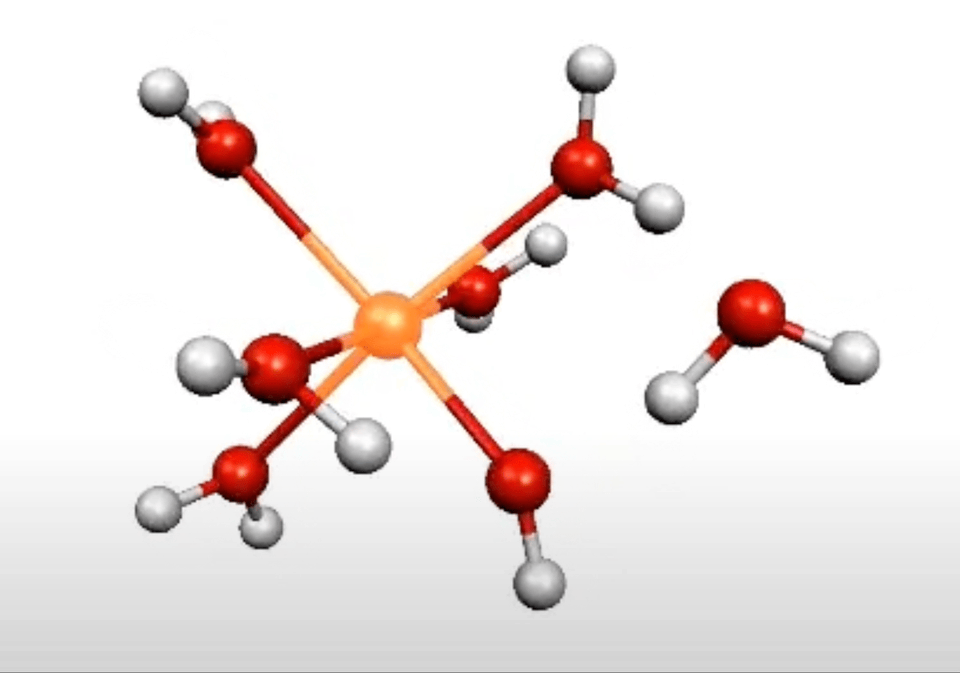
More on From Atoms To Words:
▸ All-Atom Molecular Dynamics of SARS-CoV-2: The Computational Microscope’s View of 305 Million Atoms
▸ Multiscale Simulations of DNA: From Quantum Effects To Mesoscopic Processes
▸ The Evolution of Quantum Chemistry: From Pencil and Paper to Quantum Computing
Modeling Reactivity Beyond Quantum Chemistry
As we have previously seen on From Atoms To Words, molecular dynamics is our computational microscope to peer into the atomistic dynamics of everything from DNA and proteins to the materials that make up our world.
Molecular dynamics works its magic through a set of atomic parameters known as the force field, crafted from the ground up with quantum chemistry calculations (or to match experimental values) to sketch out the energetic interactions that dictate atomic behavior.
In the classical framework of molecular dynamics, we usually simulate nonreactive scenarios—atoms held in a delicate balance by angle-strain, the gentle pull of van der Waals forces, or the stronger attraction/repulsion of Coulombic forces.
But what happens when we venture into the bustling dynamics of chemical reactions, where bonds are constantly forming and breaking? Here, traditional molecular dynamics finds its hands tied, unable to fully grasp the dynamism of chemical processes.
Enter ReaxFF, the game-changer designed to bridge this gap.
ReaxFF expands the horizons of molecular dynamics by enabling it to simulate bond breaking and forming over a range of time and space scales, vastly larger than what quantum chemistry can accommodate.
Since its pioneering introduction in 2001 for hydrocarbon reactions, ReaxFF has embarked on a remarkable journey of growth. By 2003, it had broadened its toolkit to model the chemistry of silicon, silica, and oxygen, incorporating sophisticated parameters to handle a variety of bond orders and lone-pair energies. This expansion significantly widened ReaxFF’s applicability, bringing a rich diversity of inorganic and organic systems into its fold. The story took a compelling turn in 2008 with the Chenoweth et al. study, which underscored ReaxFF’s extraordinary versatility across different elements by meticulously modeling hydrocarbon combustion.
Today, ReaxFF stands as a beacon of collaborative scientific advancement, continually evolving in tandem with the field of computational chemistry. It models the nuanced interactions of complex systems with an accuracy and depth that was once thought unattainable.
But how does ReaxFF accomplish its remarkable feats?
Further reading: The ReaxFF reactive force-field: development, applications and future directions | 2016

More on From Atoms To Words:
▸ Enzymatic Reactions: Quantum Chemistry Modeling of Life’s Catalysts
▸ Machine Learning in Materials Science: A Second Computational Revolution?
▸ Modeling the Origins of Life: Quantum Simulations of the Primordial Soup
How Does ReaxFF Molecular Dynamics Work?
At the core of ReaxFF’s force field is a clever mix of bond-order formalism and a polarizable charge model. This approach tackles the nuances of covalent and electrostatic interactions, making ReaxFF a go-to tool for modeling a vast spectrum of chemical systems.
So, how does ReaxFF really get down to business?
It begins by calculating the energy contributions from a variety of interactions within the model. We’re talking about the forces at play during bond formation, the strains in valence angles, the twists and turns of torsional angles, and even the penalties for over-coordination. Plus, ReaxFF has a knack for adding special touches to model lone-pair effects or hydrogen bonding to fit the unique characteristics of different systems.
The heart of the method lies in how ReaxFF calculates bond order. Imagine atoms getting closer or drifting apart—ReaxFF adjusts the bond strength accordingly, ensuring a smooth transition that mirrors real-life chemical processes. This approach not only keeps the simulation grounded in reality but also paves the way for modeling chemical reactions and material properties.
And here’s something cool: ReaxFF can zoom in on covalent interactions from a distance, predicting reaction barriers and handling the subtleties of transition states. This long-reach capability is key for capturing even the most fleeting covalent interactions, extending up to 5 Angstroms or beyond if needed.
In a nutshell, ReaxFF captures the essence of bond forming and breaking by dynamically adjusting bond orders in tune with atomic proximity. This method offers a clear depiction of chemical reactions, tracking the energy shifts as bonds rearrange and ensuring simulations to be as close to the real deal as possible.
Of course, a tricky part of ReaxFF-based simulations is generating the force field itself. After all, molecular dynamics is only as good as its set of parameters. But that is a story for another day.
Yet, if approached with care, ReaxFF molecular dynamics can really help a helpless chemist understand their chemical system.
Curious to see ReaxFF in action? Let’s explore some cool applications, from catalysts to complex systems in different phases.
Further reading: The ReaxFF reactive force-field: development, applications and future directions | 2016

More on From Atoms To Words:
▸ Is Machine Learning Going to Replace Computational Chemists?
▸ Water’s Hydrogen Bonds: What Makes Them Vital for Life As We Know It?
▸ Quantum Chemistry of Molecule-Surface Adsorption: The 30-Year Struggle To Chemical Accuracy
3 applications of ReaxFF molecular dynamics
Alright, let’s dive deeper into the applications of ReaxFF and uncover the magic it brings to the chemical table. We will explore how ReaxFF molecular dynamics expands our possibilities in computational chemistry, from heterogeneous catalysis to atomic layer deposition.
▸ Atomistic-level Insight | ReaxFF for Atomic Layer Deposition
Picture this: ultrathin films getting laid down super carefully, one atomic layer at a time, to hit that sweet spot of precision that’s the heartbeat of today’s semiconductors. Atomic layer deposition is a big deal in microchip manufacturing. Now, this precision is non-negotiable. Every nanometer counts.
But here’s the rub: getting a clear picture of the whole process at the atomic level is tricky. Why? Because peering into the surface states and untangling the complex chemical dynamics at play is no small feat.
ReaxFF truly shines, giving us a detailed peek into atomic layer deposition with the kind of detail that makes you feel like you’re right there watching the movie of aluminum-based molecules adsorb onto oxidized germanium surfaces—a material that brought microelectronics two Nobel Prizes. This atomic spectacle, validated by advanced spectroscopy techniques, showcases aluminum steadfast coverage as the temperature rises. On a wider note, ReaxFF molecular dynamics is becoming central to developing high-ĸ dielectric/non-Si semiconductor interfaces, setting the stage for the microchips of tomorrow. One atomic layer at a time.
▸ Versatility over large time scales | ReaxFF for Catalysis
Let’s talk nickel catalysts, shall we? These champs are key players for carbon-nanotube growth, guiding a complex process where carbon atoms dissolve into nickel nanoclusters and then dynamically reform. This nanoscale alchemy, while pivotal for advancing nanotechnology, presents significant challenges for traditional quantum chemistry due to the vast time and length scales involved.
Here, ReaxFF takes center stage. The studies conducted by Mueller and Neyts delved into carbon-nanotube formation, showing how dissociative adsorption of hydrocarbons on nickel surfaces can initiate carbon-nanotube growth.
And just like that, ReaxFF shows off its skills with oxide catalysts too. It’s pretty awesome at modeling the catalytic properties of compounds like vanadium oxide, a key player in sustainable tech and green chemistry. The ReaxFF molecular dynamics simulations by Chenoweth, for instance, investigated sulfuric acid production and pollutant removal to pinpoint the most effective catalyst structures.
▸ Reactivity across Phases | ReaxFF For Complex Systems
Regular readers of From Atoms to Words know of my fascination with graphene—from its history and remarkable properties to its myriad applications.
Now, a study that particularly captures my imagination is the work of Bagri, which leveraged ReaxFF molecular dynamics to study the defect formation in graphene oxide reduction. Their insights shed light not just on graphene-related chemical processes but also on the broader topics of metal surface oxidation and hydrogen adsorption kinetics. Such research is a cornerstone in graphene production techniques and material degradation under extreme conditions, like those faced by spacecraft heat shields.
Venturing further, the research by Achtyl on proton transfer through graphene layers, alongside Hatzell‘s investigation into the use of graphene electrodes in energy generation, underscores the versatility of ReaxFF. These studies simulate the electrical properties of graphene, consolidating its potential for renewable energy technologies.
ReaxFF isn’t just a tool for solid-state physics; its aqueous-branch parameter set has opened new frontiers in understanding aqueous chemistry and biomolecular dynamics. The valuable efforts by Rahaman on the tautomerization of glycine in water, coupled with Monti‘s extension of parameter sets to encompass amino acids and peptides, have enhanced our ability to simulate biomolecular interactions and conformational dynamics in aqueous environments.
More on From Atoms To Words:
▸ 60 Years in the Making: AlphaFold’s Historical Breakthrough in Protein Structure Prediction
▸ Large Language Models for Chemistry: Is the Beginning of a New Era?
▸ Computational Chemistry 2043: A Quantum Peep into the Future
A final personal touch
Alright, we’ve made it! Phew, that was a lot of ground to cover. We’ve seen how ReaxFF leverages a bond-order formalism to capture the dynamics of forming and breaking chemical bonds across a spectrum of materials and processes, sidestepping the hefty computational demands of quantum chemistry.
So, for those of you drawn to the chemical big picture—large models, dynamics, environmental effects, explicitly capturing pressure and temperature, but not willing to overlook the reactivity—ReaxFF molecular dynamics is your go-to tool.
Yet, the story doesn’t end here. The field is buzzing with efforts to further refine ReaxFF’s capabilities, like achieving an explicit description of electrons. These advancements aim to sharpen its precision in estimating electron affinity and extend its reach into new areas of materials science, such as the chemistry at battery interfaces and the dynamics of piezo- and ferroelectric materials.
As computational chemists, we learn to pick the right tool for each scientific challenge. And with ReaxFF molecular dynamics continuously evolving, it’s significantly enhancing our toolkit for the study of complex systems.
New discoveries, from aqueous chemistry to materials science, are now an inch closer.
If you enjoyed this dive into reaxFF molecular dynamics and its applications, I’d love to hear your thoughts. Agree, disagree, or have a totally wild theory of your own? Let’s connect! Subscribe to my LinkedIn newsletter and let’s keep the conversation rolling.