I just got back from Deloitte’s quantum climate challenge award ceremony in Hanover, Germany. And let me tell you, it was lit. The whole point of this initiative was to come up with a quantum computational design of metal-organic frameworks, aka MOF’s, all in the name of fighting climate change and saving the planet. The competitors used actual quantum computers (IBM and AWS) to test their super fancy algorithms, while the classical reference was calculated on QuantistryLab, our simulation platform. One of the questions during the discussion was: When will we achieve the so-called quantum advantage? Well, the honest answer is: nobody knows for sure. So, as simulation people, shall we sit around twiddling our thumbs waiting for quantum computing to arrive?
Here’s the good news: we already have at our disposal a powerful army of computational chemistry and quantum chemistry methods that run on classical high-performance computers. That means: we can use these simulations today to make some serious headway in the fight against climate change.
And that, dear reader, is the focus of this story. But, before digging into it, let’s talk MOF’s.
Metal Organic Frameworks To Save The Planet
Look, let’s get real for a second. I hope we agree that our emissions of CO2 are linked to some serious climate issues. It’s like, common knowledge at this point, right? That’s why we should all be fired up about finding ways to reduce greenhouse gases, especially CO2, that are getting pumped into the air.
How do we do that?
Well, first: we’ve got to cut down on our CO2 emissions – no brainer. But let’s face it, even if we do everything perfectly from here on out, it’s not going to be enough. The harsh truth is that we’re already in deep. So, we need to start figuring out how to extract that excess CO2 from the atmosphere.
Now, the ideal technology should capture CO2 selectively and convert it into useful stuff, like chemicals and fuels. This way, we not only reduce CO2 emissions but we also create chemical value to boost the energy supply for the whole world.
It’s a win-win situation.
Well, here’s the deal: that technology already exists. It’s the metal organic frameworks, aka MOF’s. These are chemical megastructures with mesmerizing periodic features. They are truly beautiful to look at.
Chemistry, for once, is a good thing – A work of art.
Further reading:
▸ Global mismatch between greenhouse gas emissions and the burden of climate change, 2016
▸ Evaluation of MIL-47(V) for CO2-Related Applications, 2013

With about 100,000+ structures available (have fun searching through the CSD), MOF’s should amaze you. Their exceptional properties surely amaze chemists.
MOF’s are made through a self-assembly process, combining metal building blocks and organic ligands to create crystalline, nanoporous structures. The exciting thing about MOF’s is that you can fine-tune their hierarchical structures, shapes, pore sizes, and surface chemistries through precise atomic-level design.
MOF’s have numerous applications such as storing and separating gases, catalysis (including CO2 conversion, water splitting, C-H bond activation), and even medical devices.
BUT here’s the catch: not all MOF’s are created equal. It’s not like you can just pick any MOF and expect it to work for what you need. Figuring out which ones are best for a given application can be a real head-scratcher. It’s like trying to find a needle in a haystack.
Plus, while MOF’s are pretty versatile, thanks to their adjustable parameters, with that versatility comes a bit of a downside. For example, MOF’s can be pretty sensitive to external conditions, like moisture. Also, their mechanical properties and catalytic capabilities can vary widely.
So, how do search for the right MOF? How do we design the best possible structures?
There are a couple of ways to go about this.
One option is to sift through the thousands of MOF structures out there, conduct a bunch of experiments, and see what happens. But that’s pretty time-consuming and not very efficient. It’s like walking in a forest and try out all possible paths to find your way.
Or, before start exploring that forest, you draw a map and reduce the number of possible paths with simulations. That’s right. Computational design of MOF’s can be a great support to identify and create the best candidates for your green purposes.
And that’s what we are talking about today – how to accurately and efficiently screen and simulate MOF structures to identify the best ones for a specific application.
So, buckle up, buttercup!
Further reading: Density functional theory–assisted calculations of structures and properties of MOFs, 2020
Computational Design of MOF’s?
One of the great things about MOF’s is their structural diversity, making them an ideal playground for computational chemistry. And indeed, computational chemistry has become an essential tool in the investigation of MOF structures, not only to complement experiments by providing detailed explanations for observed phenomena, but also to predict how certain MOF’s will behave in specific conditions, and therefore support their design.
When it comes to design, some people might envision things like sketches, blueprints, or maybe even a 3D model. But for us quantum chemists, design is all about computational modeling, density functional theory (DFT), molecular mechanics and dynamics – and all that good stuff.
With this range of methods, you can determine geometric parameters of real and hypothetical structures, such as pore size/distribution and surface area. You can estimate the stability of different MOF’s, confirm their binding capabilities, and simulate adsorption dynamics. You can investigate key processes like O2 uptake, H2 storage, CO2 capture, and climate-friendly catalytic reactions (such as CO2 conversion).
All this with the final aim of guiding the experimental intuition towards a more effective and rational design of MOF’s.
Ready to roll up your sleeves and dive into the nitty-gritty?
More on From Atoms To Words:
▸ Chemical Space to Material Discovery: Simulations and Machine Learning Leading the Way
▸ Bridging Theory and Experiment: 14 Reasons Chemical Simulations Stand as the Third Pillar of R&D
▸ Quantum Chemistry of Molecule-Surface Adsorption: The 30-Year Struggle To Chemical Accuracy
5 Recent Studies of Computational Design of MOF’s
1. DFT-based screening for Hierarchical MOF megastructures
One of the most active research areas in the field of MOF’s aims to understand how complex megastructures form from discrete molecular building blocks. MOF’s are unique in that they possess a hierarchical structure spanning multiple length scales, ranging from nanoparticles to 2D sheets and thin films, before culminating in the creation of marvelous 3D architectures.
To shed light on this extraordinary process, Kwon & Co. employed a DFT-based screening method to examine a whopping 9,484 MOF building blocks. By investigating how the metal nodes of one MOF coordinates with the linkers of another MOF, they identified six MOF pairs that could seamlessly connect with one another.
And guess what? The predicted structures were successfully synthesized, resulting in clean single crystalline MOF@MOF structures.
The work of Kwon & Co. not only offers insights into the fundamental processes underlying MOF megastructure formation, but also confirms that the combination of computationally generated knowledge and experimental techniques can help fabricate a broader range of MOF structures suitable for specific applications.
Now, that’s what I call a chemist dream come true!

2. Computational Design of moisture-stable MOF’s
As we have seen so far, MOF’s have enormous potential in a wide range of applications, but their unique porous structure and chemical versatility come at a cost: precarious stability to moisture. And that’s a problem that hinders their widespread use in the fight against climate change.
But fear not! With the support of molecular mechanics, grand canonical Monte Carlo, and DFT, Chen & Co. designed two new MOF’s from the original MIL-88 type MOF by modifying it with different types of functional brackets. The result? They synthesized the first examples of moisture-stable MOF’s that also have a high acetylene (C2H2) uptake capacity.
Not only that. Thanks to a combination of experimental ingenuity and computational design of MOF’s, Chen & Co. also demonstrated how the framework stability and storage capacity of these systems can be optimized by modulating geometrical parameters, such as pore size and distribution.

More on From Atoms To Words:
▸ Computational Chemistry 2043: A Quantum Peep into the Future
▸ Do We Really Need Quantum Computing in Chemical R&D?
▸ All-Atom Molecular Dynamics of SARS-CoV-2: The Computational Microscope’s View of 305 Million Atoms
3. Multiscale computational screening of MOF mechanical landscape
So, yes: MOF’s are incredibly versatile materials, but their adoption in industry is still limited by some fundamental issues. Many MOF’s are vulnerable to loss of crystallinity, due to chemical reactions or mechanical stress. This can lead to pore collapse and degradation of their amazing abilities.
But there’s hope on the horizon! In a beautiful and dense study, Moghadam & Co. employed a thorough multi-scale modeling approach to map out the structure-mechanical landscape of MOF’s.
They started by using high-throughput molecular simulations on a massive dataset of 3,385 MOF’s and 41 distinct network topologies. Then, they developed a machine-learning algorithm to predict the mechanical properties of MOF’s. Finally, they conducted molecular dynamics simulations to pinpoint the loss of crystallinity within a given topology.
Through their analysis, Moghadam & Co. revealed that the mechanical properties of MOF’s are highly sensitive to a few key structural parameters such as topology, coordination characteristics, and building block nature.
Amazingly, Moghadam & Co made their multiscale screening tool available in the hope that it may help future research accelerate the translation of MOF’s to industrial applications.
Here’s the trailer:

4. Fully predictive simulations of CO2 capture
With their open metal sites, MOF’s have the potential to efficiently capture CO2 and reduce it to CO or separate it from other gas mixtures. However, figuring out how guest molecules interact with these open metal sites is no easy feat.
To address this challenge, Becker & Co. employed quantum chemistry calculations to develop a fully predictive polarizable force field. With this new shiny tool, they were able to describe the adsorption process of different MOF structures by means of computational modeling alone.
This means that their tool can speed up the selection process of the most suitable MOF for specific applications without relying on costly and laborious experimental work.
The future of CO2 capture just got a whole lot brighter.
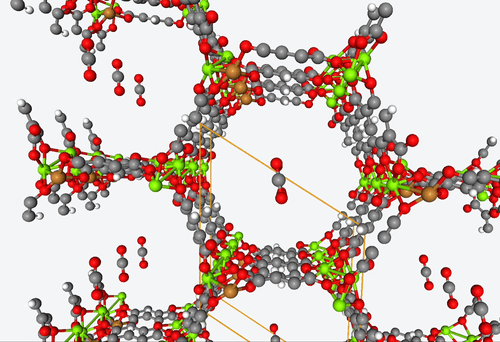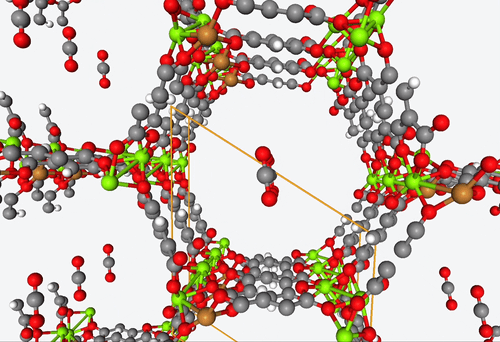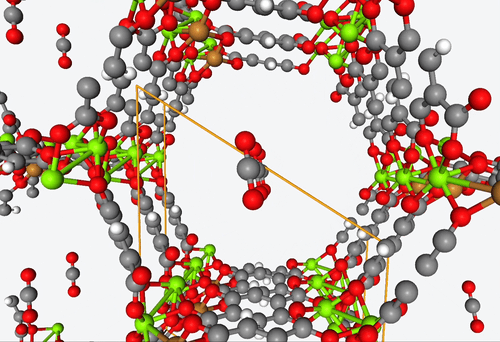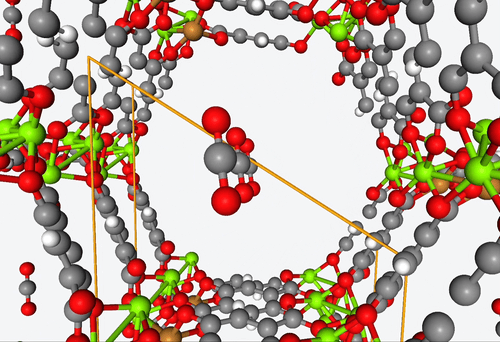
5. DFT-based high-throughput screening of catalytic MOF’s
MOF structures containing open metal sites are ideal for facilitating catalytic processes like CO2 reduction, water splitting, and C-H bond activation.
Let’s take C-H bond activation for example, a fundamental process for molecular syntheses, with applications in pharmaceuticals and material sciences, particularly for the conversion of methane to methanol.
With this specific focus in mind, Rosen & Co. developed and employed a fully automated DFT-based high-throughput workflow to screen promising MOF candidates. Their approach paid off with critical insight into the characteristics of the ideal MOF for heterogeneous catalysis and C-H bond activation.
One of the conclusions drawn from these simulations is that the metal oxidation step, rather than the activation of the C–H bond in methane, corresponds to the largest energetic barrier. This means that the development of new MOF’s should focus on low-valence, redox-active open metal sites.
Perhaps most importantly, Rosen & Co. confirmed that DFT-based high-throughput workflows can be a powerful tool for the design of MOF’s with unique physicochemical properties, which may maximize desired catalytic potential for CO2 reduction, water splitting, and C-H bond activation.
More on From Atoms To Words:
▸ Large Language Models for Chemistry: Is the Beginning of a New Era?
▸ The Evolution of Quantum Chemistry: From Pencil and Paper to Quantum Computing
▸ When Will RNA Structure Prediction Get Its AlphaFold Breakthrough?
A Final Personal Touch
So, let’s circle up to the beginning. The Quantum Climate Challenge award ceremony was pretty damn awesome. I mean, I met some seriously cool people and I gotta say, it left me feeling a little more hopeful about the future of our world, you know what I mean? And not just that, but the future of computational and quantum chemistry and computational design of MOF’s.
Some of the ideas for quantum solutions to investigate and design MOF’s were seriously slick. But, let’s not get ahead of ourselves here. There’s still a whole lot of room for improvement and innovation in this field. And let’s be real, we’re talking about quantum computing here – who knows how long it will take, 5, 10, 15 years?
But, while we’re dreaming up the ultimate holy-grail combo of quantum chemistry on quantum computers, let’s not forget about the computational potential we have at our fingertips right now.
So, let’s start simulating today to save the world tomorrow. It’s not just possible, it’s useful and it damn well works.
If you enjoyed this dive into metal-organic frameworks and their amazing properties, I’d love to hear your thoughts. Agree, disagree, or have a totally wild theory of your own? Let’s connect! Subscribe to my LinkedIn newsletter and let’s keep the conversation rolling.